中国医药市场持续增长,是仅次于美国的全球第二大医药产业。预计到 2023 年,中国医药市场规模将达到 1,618 亿美元,未来几年平均增长率为 5%,占全球市场份额的 30%。
新药准入的加速、单品类监管格局的变化、中国社会的快速老龄化以及随之而来的日益增长的医疗需求,都为医药市场提供了一系列机遇。
13 年后,中国监管部门对药品注册规则和指南进行了多次修改。这些变化旨在降低复杂性,并与全球其他监管机构保持一致。
中国药品注册新规定的亮点和机遇
让我们来看看中国监管机构新规定的一些亮点
--按照全球法规,CTA 审批时限为 60 天。
--明确不同类型药品注册申请的审评审批时限。
--简化药品注册和生产许可之间的流程,确保 GMP。
--改善申请人与药物评价中心(CDE)之间的沟通,以实现有效的 CTA 流程。
--引入与技术审查并行的基于风险的现场检查和实验室测试注册。
--临床试验阶段的突破性审批和有条件审批,以及上市审批阶段的优先审评,加快了药品注册速度,满足了中国尚未满足的医疗需求。
中国药品注册管理机构
国家医药管理局(NMPA)是负责药品注册管理、制定药品注册规范、组织药品注册审评审批的主要监管部门。
国家医药管理局药品审评中心(CDE)负责药品临床试验申请、药品上市许可申请、补充申请和境外生产药品再注册申请的审评。
哪些人可以在中国申请药品注册?
上市许可持有人(MAH)可以提交药物临床试验申请、药物上市许可申请、重新注册申请和补充申请。除这些职责外,MAH 还可以处理其他各种职责。
中国的药品注册类别
在中国开发和注册药品的要求取决于进一步的分类:
--化学药
--生物制品
--传统中药。
每种类型又分为三个注册类别,这决定了申请人在申请注册时必须提供的材料,例如临床试验申请、上市许可申请等。
化学药的注册类别如下
--创新药物
--改进型新药
--仿制药
对于生物制品,注册类别如下
--创新生物制品
--经改进的新生物制品
--已上市生物制品(包括生物仿制药)
相应的申请材料要求是根据注册药品的产品特性、创新程度和审评管理需要确定的。
申请人选择注册的药品类别决定了临床试验申请的审评审批程序。NMPA 负责管理临床试验申请。
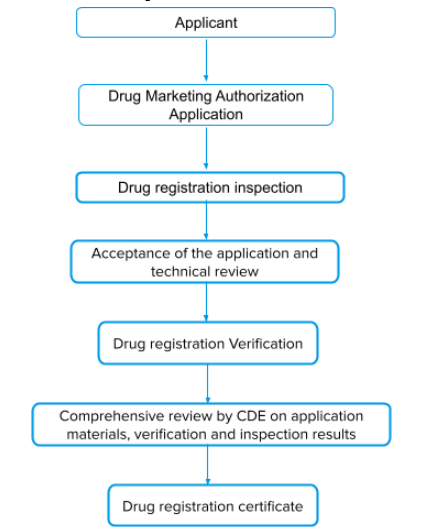
药品上市许可的审查程序
--在完成支持药品注册的临床前研究和临床研究后,申请人应按照要求提交药品上市许可申请。
--对申请材料进行形式审查,符合要求的予以受理。
--药品审评中心(CDE)组织药学、医学等技术人员对受理的药品上市许可申请进行审评。
--综合审评结束后,颁发药品注册证。综合审评结论未通过的,不予批准。
药品注册证的有效性和重新注册
药品上市许可持有人的药品注册证有效期为五年。在有效期内,药品上市许可持有人必须对上市药品的安全性、有效性和质量可控性负责,并在有效期满前六个月申请药品再注册。
药品注册的加速审批
根据相关规定,国家药品监督管理局(NMPA)启动了新药审评流程,并将审批流程分为以下四类:
--突破性审批
--优先审评
--有条件批准
--特殊批准
--申请人需明确这些审评流程适用于哪一类药物,以及在药物开发的哪个阶段可适用。
审查和批准登记费
申请人需根据适用法规支付费用。作为药品注册程序的一部分,国家医药产品管理局(NMPA)收取药品注册费,用于审查和批准临床试验。不同类别的药品收费如下。
--国产新药
--中国境外生产的新药
--中国生产的非专利药品
--中国境外生产的非专利药品














