自 100 年前胰岛素问世以来,已有 80 多种多肽药物上市,用于治疗糖尿病、癌症、骨质疏松症、多发性硬化症、艾滋病和慢性疼痛等多种疾病。多肽作为后基因时代的热门话题,已被广泛应用于药物开发。自然-药物发现评论》(Nature Review Drug Discovery)最近发表了一篇题为 “多肽药物开发趋势 ”的综述,总结了多肽药物开发的主要趋势。
多肽药物开发的里程碑
1922 年,第一种多肽药物胰岛素问世,这种药物最初是从牛和猪的胰腺中提取的,用于治疗糖尿病。1954 年,化学家 Vincent du Vigneaud 首次完成了催产素和血管加压素的全合成,并因此获得 1955 年诺贝尔化学奖。1963 年,固相多肽合成(SPPS)技术诞生,通过在固相上组装氨基酸,实现了多肽合成的自动化,极大地促进了多肽药物的开发。20 世纪 80 年代重组技术的出现,使大规模生产绿色多肽成为可能。肽类药物最大的缺点是血浆半衰期短,容易被肾脏滤过清除,近年来人们开发了通过与脂质、蛋白质或聚乙二醇(PEG)共价连接来增加肽分子量的策略,以克服这一缺点。新的筛选技术和新的化学方法也促进了多肽药物的开发(图 1)。
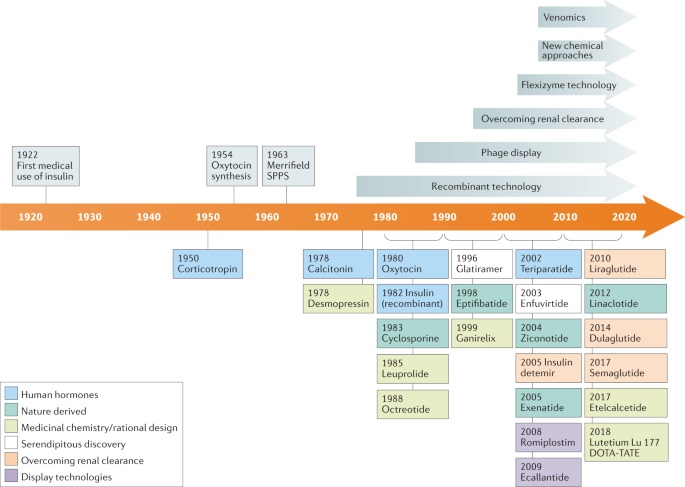
多肽药物市场
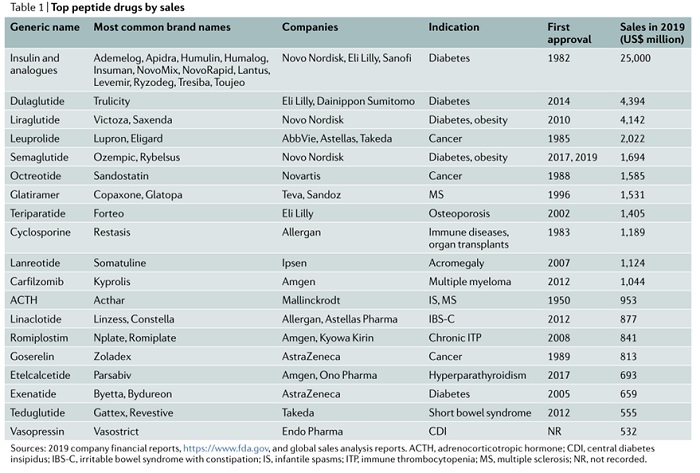
多肽药物填补了小分子化学药物和大分子生物制剂之间的空白,占据了特殊的地位。过去 60 年间,全球获批的多肽药物数量稳步增长,平均增长率为 7.7%(图 2b)。2019 年,多肽药物的全球销售额超过 500 亿美元,占全球医药市场的 5%(图 2a),其中胰岛素及其类似物约占 50%(250 亿美元),其次是胰高血糖素样肽 1 (GLP1 ) 受体激动剂度拉鲁肽(44 亿美元)和利拉鲁肽(41 亿美元)(表 1)。目前获批的多肽治疗药物大多是激动剂(图 2c),最常见的适应症与内分泌、代谢和肿瘤有关(图 2d)。

肽类激素的研发
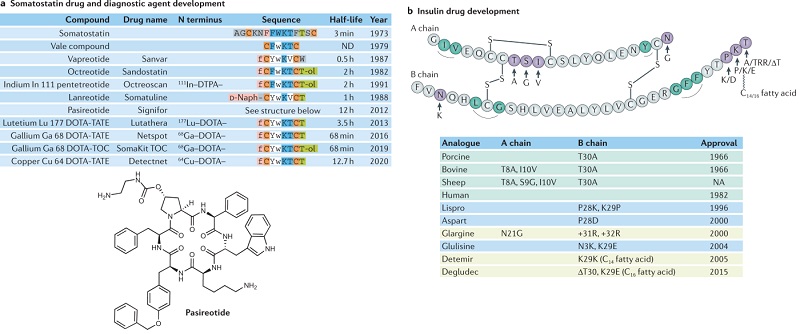
二十世纪初的肽类药物研究主要集中在人体信号激素的作用上。内源性多肽激素的半衰期很短,只有几分钟,这阻碍了它们的临床应用。科学家们采用多种药物化学方法对肽进行修饰,以增加其稳定性,同时改善其他特性。其中两个具有代表性的例子是生长抑素体生长抑素和胰岛素,通过点突变、N 端或 C 端延伸、肽链环化或长链脂族烃的连接等化学修饰,提高了这些内源性配体的稳定性、效力和选择性,最终有几种药物成功上市(图 3)。
天然肽
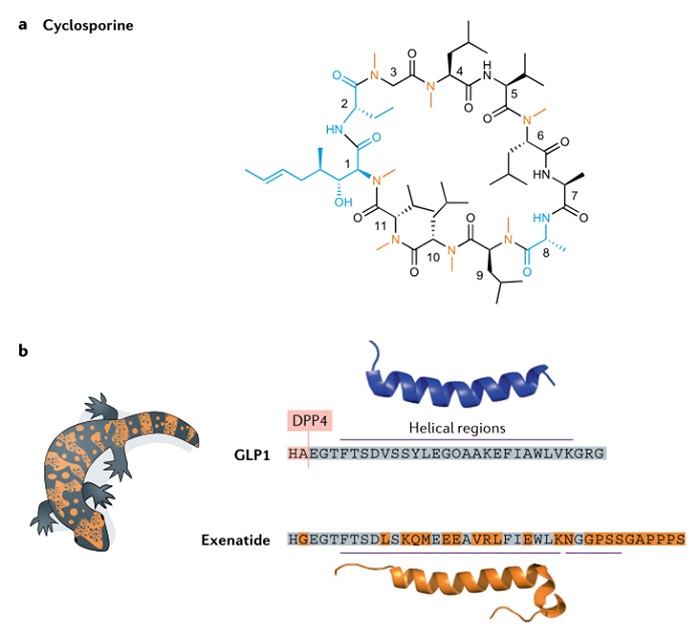
环孢素(图 4)是从真菌中分离出来的一种多肽,是一种中性、疏水性环肽,含有 11 个残基。环孢素的高度氮甲基化和环状结构使其具有抗蛋白酶水解的能力,此外,疏水性和构象灵活性使其具有口服生物利用度。环孢素作为一种免疫抑制剂于 1983 年获得批准,这是发现天然产品药用价值的一个鼓舞人心的例子,同时也提醒人们肽和肽类似物作为口服药物开发的潜力。艾塞那肽(图 4b)提取自剧毒蜥蜴的毒液,于 2005 年获批用于治疗 2 型糖尿病,是第一个非常成功的 GLP1 受体激动剂药物。人 GLP1(7-37)的血浆半衰期很短,会被二肽基肽酶 4(DPP4)迅速降解,并在 1-2 分钟内被肾脏排出体外。艾塞那肽与人类 GLP1(7-37) 的同源性为 53%,是一种完全的 GLP1 受体激动剂,具有抗 DPP4 降解的稳定性和较低的肾脏消除率。
多肽药物发现策略
毒液组学和显示技术是发现治疗肽的两项关键技术。毒液组学利用生物信息学分析来自有毒动物的基因组和转录组数据,以及从粗毒液样本中获得的蛋白质组数据。这种方法可以识别大量毒液肽序列,然后通过合成或重组技术获得这些序列,并针对治疗靶点进行筛选。显示技术,包括噬菌体显示、酵母显示、mRNA显示、核糖体显示和脱氧核糖核酸显示,是当今更先进的多肽药物发现技术之一(图5)。它们在表型(多肽)和基因型(DNA 或 RNA)之间建立了联系,可以生成样本量巨大(1010-1015)的多肽库,并针对治疗靶点进行筛选。经过多轮筛选获得高亲和性多肽先导物后,再利用药物化学策略改善先导物的成药特性。
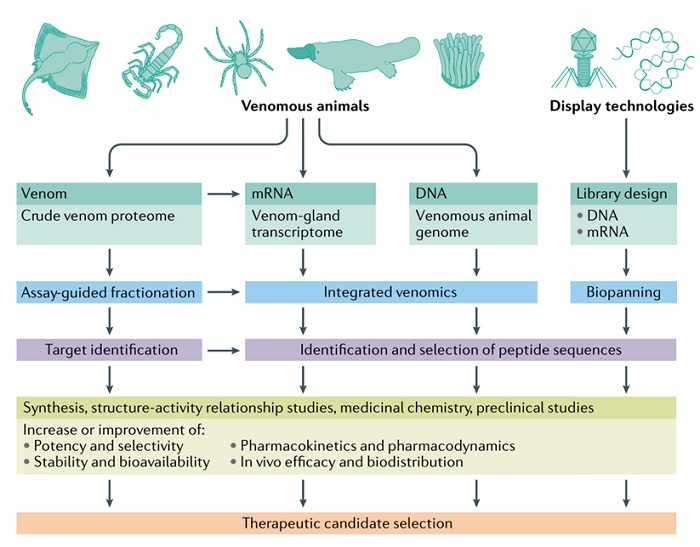
多肽药物的药物化学战略
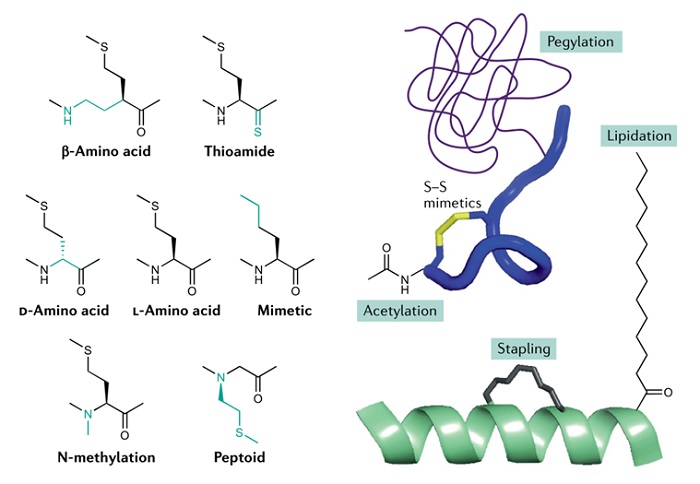
传统的药物化学策略,包括 N 端乙酰化、N-甲基化、使用 d-氨基酸、非天然氨基酸和酰胺键模拟物(如硫代酰胺、模拟肽和β-氨基酸)(图 6),在调节多肽代谢稳定性和生物利用度方面简单有效,将继续在多肽药物开发中发挥不可替代的作用。新的化学修饰方法,如 N 端到 C 端环化、使用二硫键模拟物、α-螺旋钉固化和缀合肽等,将越来越多地用于多肽药物的设计,以提高药物的可药性。
多肽给药技术的进展
大多数肽类药物都是通过注射给药,这种给药方式存在病人依从性差、感染风险大、针头废弃物具有生物危害性等缺点。因此,开发注射给药的替代品已成为当务之急。各种泵技术提供了注射给药的替代品,如基于微技术的植入式泵,它能更精确地控制给药,但泵的植入是侵入性的,使用时需要重新注药。非注射给药替代方法包括无针注射,如液体喷射注射器,但注射部位可能会出现疼痛和出血,给药量也无法精确控制。其他方法包括使用皮肤穿透肽、超声波或电场来增加肽类药物的皮肤穿透力。肺部和鼻腔给药是另外两种潜在的给药途径,药物已经上市,但存在安全问题,给药效率有限。此外,在开发多肽药物的口服给药策略方面也投入了大量精力,但口服给药受到多肽在胃肠道中酶消化和在肠上皮细胞中渗透有限的限制。克服这些障碍的一种策略是将多肽与渗透增强剂结合使用,这种策略在临床上取得了成功(如塞马鲁肽)。
其他策略包括调节 pH 值、使用粘液渗透剂、酶抑制剂、水凝胶、肠贴剂、肠胶囊或肠装置。细胞内蛋白质-蛋白质相互作用(PPI)也是重要的药物靶点,鉴于大多数肽具有较低的膜渗透性,人们还开发了细胞穿透肽(CPPs),以促进肽类药物跨细胞膜递送。
多肽药物开发的前景
总之,多肽药物开发领域在过去 60 年中克服了许多关键挑战,但仍有很大的改进和发展空间。尽管在给药技术、制剂技术和药物化学方面取得了巨大进步,但 90% 的多肽药物仍通过注射给药,缺乏口服生物利用度仍是限制多肽药物开发的主要因素。高昂的研发成本和无法实现的收入预期是多肽药物开发的另一个重要障碍。尽管存在这些众所周知的障碍,多肽药物市场仍在稳步增长,因此,选择适当的治疗领域对多肽药物开发至关重要。














